30. Medfødt hjertesygdom


30.1 Forekomst
Medfødt hjertesygdom (CHD) forekommer hos ca. 0,8% af alle levendefødte børn. Mere end 85% overlever til voksenalderen, hvilket svarer til en årlig tilgang af 400-500 voksne patienter med såkaldt ACHD (Adult Congenital Heart Disease). Denne gruppe udgør mere end 20.000 patienter i Danmark. Langtidsprognosen for de fleste simple medfødte hjertefejl er god, men der er fortsat nedsat overlevelse i forhold til baggrundsbefolkningen. De fleste komplekse hjertefejl har betydeligt nedsat overlevelse, og mange har brug for løbende interventioner og operationer. På grund af forbedret prænatal diagnostik og mulighed for sen-abort er incidensen af de mest komplekse medfødte hjertesygdomme faldende.
30.2 Organisationsstruktur for behandling og opfølgning af patienter med medfødt hjertesygdom i Danmark
Invasiv og kirurgisk behandling af børn og voksne med medfødte hjertesygdomme er en højt specialiseret funktion, som varetages på Rigshospitalet.
Opfølgning og udredning af børn og voksne med CHD er ligeledes en højt specialiseret funktion, der varetages af højt specialiserede enheder (HSE), som i Danmark er fordelt mellem Aarhus Universitetshospital (AUH), Odense Universitetshospital (OUH) og Rigshospitalet (RH). De fleste CHD patienter skal følges livslangt. Patienter med kompleks CHD følges primært på AUH, OUH og RH. Patienter med mindre kompleks CHD følges på udvalgte større kardiologiske afdelinger af speciallæger med særlig uddannelse og interesse indenfor CHD. Dette foregår i et tæt og formaliseret samarbejde med en af de tre HSE; AUH, OUH og RH. De tre HSE er organiseret med anæstesiologisk- og kardiologisk specialviden omkring den perioperative håndtering af CHD-patienter. Implantation af pacemaker og ICD-enheder samt ablation foretages på alle HSE, men ved kompleks CHD kun på RH.
30.3 Akutte og generelle problemstillinger
Det er forholdsvis sjældent, at ACHD præsenterer sig med akutte medicinske problemstillinger. En stigning i akutte kontakter må dog forventes som et resultat af den voksende og aldrende ACHD-population. De mest almindelige akutte præsentationer er akut hjertesvigt og arytmi (2/3 af akutte kontakter). Som udgangspunkt skal ACHD-patienter behandles som alle andre patienter med akut sygdom.
Generelt:
- Spørg til vanlig saturation ved cyanose. Denne er sjældent akut opstået, men kan være forværret ved cyanotisk CHD.
- Mål blodtryk på højre arm men gerne begge arme til sammenligning (obs coarctatio (CoA) opereret patient).
- Husk luftfilter på perifer vene kateter (PVK) ved cyanotisk CHD (grundet risiko for luftembolier). Brug filter ved tvivl om diagnosen og hvis filter ikke haves da anvend ’våd mod våd teknik’ (tilbageløb af blod i venflon ved tilslutning af infusion/medicin så luftbobler ikke kommer ind i blodbanen).
- Grundig anamnese og objektiv undersøgelse er vigtig, herunder observation af tidligere thoraxkirurgiske cikatricer.
Vedrørende specifikke problemstillinger anbefales følgende:
30.3.1 Arytmi/synkope
- Vanlig akut DC-konvertering ved takyarytmi og hæmodynamisk instabilitet (tegn på hjertesvigt, synkope, brystsmerter eller shock).
- Hyppigste arytmi er supraventrikulær takykardi (SVT). Komplekse patienter kan blive hæmodynamisk ustabile selv af langsom SVT), specielt patienter korrigeret med Total Cavo Pulmonal Connection (TCPC) og Mustard/Senning (se Afsnit 30.10.1)).
- Arvæv efter tidligere hjertekirurgi øger risikoen for arytmi. Ved synkope og nytilkommen arytmi, indlægges patienten og konfereres på lav tærskel med en af de tre HSE.
- Hos patienter med tolerabel SVT og hvor det tidligere ikke har været muligt at fremkalde arytmien under elektrofysiologisk undersøgelse, kan det være hensigtsmæssigt at overflytte patienten til subakut ablation på HSE under pågående arytmi. I disse tilfælde tilrådes kontakt til HSE før DC-konvertering eller medicinsk behandling.
Forekomst af SVT er associeret med dårligere prognose hos patienter med moderat og svær CHD. Som udgangspunkt tilstræbes derfor rytmeregulerende strategi. Medicinsk antiarytmisk behandling anbefales i første omgang at være betablokade og amiodaron.
Henvisning til radiofrekvensablation bør overvejes hos alle patienter med betydende arytmi, særligt hvis længerevarende behandling med amiodaron er nødvendig. Calciumantagonister bør undgås pga. de strukturelle forandringer og høj forekomst af abnormaliteter i His-Purkinje systemet, ligesom flecainid er relativt kontraindiceret til kongenitte patienter.
Generelt anbefales pacemaker samt primær/sekundær profylaktisk ICD-implantation på samme indikationer som hos patienter uden CHD. Særlige tilfælde inkluderer bl.a. patienter med Fallots tetralogi (ToF) og systemisk højre ventrikel (RV). Indikation for ICD hos patienter med moderat til svær kompleks medfødt hjertesygdom skal altid konfereres med HSE
30.3.2 Hjertesvigt/akut dyspnø
- Vanddrivende behandling ved akut hjertesvigt; behandles i øvrigt som vanligt (se Kapitel 4: Akut hjertesvigt og Kapitel 5: Kronisk hjertesvigt). Patienten indlægges og konferences med HSE med henblik på evt. overflytning.
- Der tilrådes forsigtighed ved behandling med systemisk vasodilatorer til nogle af de komplekse tilstande, ved tvivl kontaktes HSE.
- Vær opmærksom på udløsende årsag (arytmi, infektion, svigtende ventrikel funktion).
30.3.3 Infektion/endokarditis
- Patienter med CHD udgør en speciel risikogruppe i forhold til infektiøs endokarditis (IE), da mange har væsentlige strukturelle forhold i hjertet, som disponerer for IE, herunder dysplastiske klapper, residualdefekter og indopererede fremmedlegemer, såsom kunstige hjerteklapper og elektroder.
- Alle pulmonal klapproteser kan udgøre fokus for IE, men tilstanden ses hyppigere hos patienter med stentklapper i pulmonal position (Melody- Venus eller Sapien-klapper), som ofte forekommer flere år efter indsættelsen. En pludseligt øget antegrad gradient over en stentklap bør give anledning til akut udredning for IE, da der i alvorlige tilfælde kan opstå hurtigt progredierende og svær obstruktion af klappen. Udredning på mistanke om IE bør foregå på HSE, gerne med transoesophageal ekkokardiografi (TEE) og/eller intrakardiel ekkokardiografi (ICE).
- Cyanotisk hjertesygdom er en selvstændig risikofaktor for IE.
- Bloddyrkninger bør sikres forud for iværksættelse af antibiotisk behandling.
- Profylakse mod IE følger generelle retningslinier (se Kapitel 7: Infektiøs endokarditis, Afsnit 7.11: Profylakse mod infektiøs endokarditis). For alle patienter anbefales god mundhygiejne og regelmæssige tandlægebesøg.
30.3.4 Brystsmerter/iskæmi/tromboemboli/aortadissektion
- ACHD-patienter er som den almene befolkning i risiko for at få iskæmisk hjertesygdom. Vær opmærksom på at patienter med intrakardielle shunts/fenestre kan få embolier til koronarkar ved manglende/dysreguleret antikoagulations (AK) behandling.
Patienter med bindevævssygdomme herunder Marfan, Turner, vaskulær Ehlers-Danlos og Loeys-Dietz syndrom er i øget risiko for aortadissektion (Kapitel 11: Aortasygdom).
- CAVE nitrater og lignende kardilatation ved Eisenmengers syndrom pga. risiko for kredsløbskollaps
30.3.5 Anæstesi
Patienter med simple hjertefejl kan som udgangspunkt bedøves og opereres lokalt efter vanlige retningslinjer (se Kapitel 24: Kardiel risikovurdering forud for ikke-hjerterelateret kirurgi).
Ved kompleks CHD afhænger risikoen ved anæstesi og operation af underliggende anatomi, ventrikelfunktion, klapproblemer og resterende shunts. Der bør altid konfereres med HSE.
Ved cyanotisk hjertesygdom/Eisenmengers syndrom bør procedurer (inklusiv DC-konvertering) foregå på HSE grundet risiko for perifer vasodilatation ved bedøvelse, som kan være fatalt.
Se endvidere Afsnit 30.13.
30.4 Abnormt anlagte koronararterier
Koronararterie anomali kan forekomme som en del af mere kompleks CHD men kan også forekomme isoleret hos voksne patienter (
Abnorm afgang af koronararterie fra pulmonal arterien
Forekommer hos mindre end et barn årligt i Danmark. Venstre (ALCAPA) eller ekstremt sjældent højre (ARCAPA) koronararterie afgår fra a. pulmonalis. Dette kan ikke erkendes ved fosterscreening. Der udvikles som regel hjertesvigt i de første levemåneder, når pulmonal trykket falder og koronar perfusionen bliver afhængig af kollateraler. Hos et fåtal er kollateral-forsyningen veludtalt, og symptomdebut ses først senere i livet, typisk i form af hjertestop eller hjertesvigt i voksen alder. Behandlingen er reimplantation af den abnormt afgående koronararterie til aorta. Hos voksne med hjertestop gives sekundær profylaktisk ICD.
Abnorm afgang af koronararterie fra aorta
Afgang af en koronararterie fra modsatte sinus er ikke i sig selv et afgørende patologisk fund. Her skal risikoen for iskæmi vurderes. Den foretrukne anatomiske billedmodalitet er hjerte-CT, der suppleres med funktionel iskæmitest under fysiologisk belastning.
Kirurgisk behandling anbefales ved symptomer på iskæmi og påviselig iskæmi i det afficerede koronararterie forsyningsområde eller højrisiko anatomi (afgang højt på aorta (>1cm fra sinotubulære overgang), slidsformet ostium eller intramuralt forløb). Behandling overvejes hos unge (
Koronararteriefistler
Fistel fra koronararterie som regel til højre atrie, RV eller pulmonalarterien er, hvis lille, uden prognostisk betydning. Større fistler kan give symptomer relateret til sværhedsgraden af volumenbelastningen, eller koronar steal. Voksne patienter kan præsentere sig med brystsmerter, dyspnø, arytmi eller myokardieinfarkt. Intervention tilbydes med kateterbaseret lukning, eller kirurgi i udvalgte tilfælde.
30.5 Septumdefekter
30.5.1 Atrieseptumdefekt (ASD)
Definition og inddeling:
ASD er en medfødt hjertesygdom, der ofte først diagnosticeres i voksenalderen, og inddeles som følger:
- Sekundum defekt (80%).
- Primum defekt (eller partiel atrioventrikulær (AV) septumdefekt) (15%). Selvstændig sygdomsenhed med atrial kommunikation på baggrund af én fælles AV-klap (se Afsnit 30.5.4). Mitral- og tricuspidalklappen vil derfor ofte være malformerede med en grad af insufficiens (”cleft”).
- Sinus venosus defekt (5%):
- Vena cava superior type er den hyppigste (90%) og er oftest associeret med partiel abnorm indmunding af højresidige lungevener (kan anatomisk anskues som en kommunikation mellem øvre højre lungevene og vena cava superior, der tillader shunt mellem atrierne (se afsnittet herunder)
- Vena cava inferior (10%) med eller uden abnormt indmundende nedre højre lungevene.
- Sinus coronarius defekt (
Hæmodynamik: Graden af venstre-højre shunt afhænger af forskellen i compliance (diastolisk funktion) af ventriklerne. Venstre ventrikel (LV) får med alderen relativt tiltagende nedsat compliance, hvilket medfører øget venstre-højre shunt.
Symptomer: Sædvanligvis ingen eller sparsomme symptomer indtil 40-50 års alderen, hvor typiske debutsymptomer er funktionsdyspnø, atrial takyarytmi, recidiverende pneumonier samt paradoks emboli (sjældent). Ved sinus venosus ASD kan der forekomme cyanose pga. strømning af systemvene blod til venstre atrium. Udvikling af Eisenmengers syndrom er sjældent.
Arytmi: Incidensen af arytmi synes at aftage efter lukning af ASD hos børn og unge, hvorfor ASD-lukning alene anbefales hos denne gruppe. Ved lukning i voksenalder ses ikke samme reduktion. Hos patienter med stor arytmi byrde og hæmodynamisk betydende ASD, bør ablation overvejes før ASD-lukning.
Behandling: Indikationen for lukning af ASD er volumenbetinget dilatation af de højresidige kamre, ratio af flow gennem det systemiske og det pulmonale kredsløb (Qp/Qs) >1,5, symptomer eller paradoks emboli.
De fleste sekundum ASD’er (også multifenestrerede) kan lukkes kateterbaseret (kræver septumkanter på 4-5 mm, men ikke nødvendigvis mod aorta). Da ASD’er sjældent giver symptomer i barnealderen, foretages kateterbaseret lukning af større defekter typisk efter fireårsalderen. Kateterbaseret lukning af store symptomgivende defekter kan efter individuelt skøn foretages før fireårsalderen.
Øvrige ASD’er lukkes vanligvis operativt, men nye stents har gjort det muligt at lukke sinus venosus defekter kateterbaseret. Trombocythæmmer og endokarditisprofylakse gives efterfølgende.
Prognose: God prognose efter lukning. Langtidsfølger er sjældne:
- supraventrikulær arytmi (se Holdningspapir om håndtering af takyarytmi ved kongenit hjertesygdom)
- device erosion (sjældent)
- pulmonal hypertension (sjældent)
- patienter med vedvarende dilatation af højresidige kamre følges med lange intervaller (3-5 år)
30.5.2 Partielt abnormt indmundende lungevener (PAPVD)
Definition og inddeling: Én eller flere, men ikke alle lungevener, drænerer direkte eller indirekte til højre atrium. Der findes mange forskellige typer. Ved de hyppigste PAPVD drænerer lungevener fra højre over- og mellemlap enten direkte i v. cava superior eller i højre atrium. Alle højresidige lungevener kan drænere til v. cava inferior (Scimitar syndrom), hvilket ofte er ledsaget af højresidige lungesekvester med sekvesterarterie fra aorta. Venstresidige lungevener kan munde ud i brovenen (v. brachiocephalicus) eller sinus coronarius. Nedre højresidige lungevener kan drænere til cava inferior (evt. infra-diagfragmatisk). Ved ASD som følge af sinus venosus defekter mod v. cava superior, ses typisk abnorm indmunding af øvre højre lungevene.
Symptomer: Isolerede tilfælde er oftest asymptomatiske i barnealderen. De hæmodynamiske konsekvenser er som beskrevet for ASD, og kan medføre dyspnø og atrieflimren i voksenalderen. Lungesekvester kan vise sig ved recidiverende lungeinfektioner og hæmoptyser.
Behandling: Ved betydende arteriovenøse shunts (dilatation af højresidige hjertekamre og/eller symptomer) foretages operativ korrektion med reetablering af forbindelsen mellem lungevenerne og venstre atrium gennem tunnelering. En enkelt abnormt indmundende vene kræver sjældent behandling. Lungesekvestre opereres med resektion og/eller kateterbaseret okklusion af den abnorme fødearterie.
Langtidsfølger: Langtidsresultaterne er sædvanligvis gode.
- ikke-opererede patienter kan i sjældne tilfælde udvikle pulmonal hypertension, højresidigt hjertesvigt og arytmier
- opererede patienter følges med henblik på udvikling af obstruktion svarende til anastomosen/tunnelen til venstre atrium og af v. cava superior (ved sinus venosus defekter med PAPVD)
30.5.3 Ventrikelseptumdefekt (VSD)
Definition og inddeling:
VSD er udbredt forekommende som del i komplekse hjertesygdomme. Her omtales solitært forekommende/tilbageværende VSD.
- perimembranøse (80%)
- muskulære (15%)
- doubly committed subarterial (5%)
Symptomer og objektive fund: holosystolisk kraftig mislyd langs venstre sternalrand. Kliniske symptomer afhænger af defektens størrelse og lungekarmodstand (PVR) og kan variere fra ingen symptomer til hjertesvigt. Børn med store defekter kan være uden mislyd og asymptomatiske, hvis PVR forbliver høj og graden af shunt dermed beskeden. Dette kan dog føre til Eisenmengers syndrom.
Behandling: de fleste defekter diagnosticeres og opereres eller lukker sig spontant i barnealderen. VSD lukkes i voksenalderen, hvis shunten findes hæmodynamisk betydende (dilatation af venstresidige kamre, aftagende LV-funktion eller RV-trykbelastning, prolaps af aortacusp med tiltagende aortainsufficiens, udvikling af obstruktion af højre ventrikels udløbsdel (RVOT) eller IE). De fleste muskulære defekter og en del perimembranøse defekter kan lukkes kateterbaseret, når der ikke samtidig er indikation for anden kirurgisk behandling. Mindre (restriktive) defekter kan håndteres afventende, og spontanlukning forekommer hyppigt.
Prognose: God, med normal livslængde og livskvalitet hos tidligt velopererede. Langtidsfølger er sjældne hos tidligt velopererede:
- inkomplet eller komplet højresidigt grenblok ses hos op til 40%, sjældnere ses AV-blok
- restdefekter kan være betydende og medføre hjertesvigt
- aortainsufficiens ved defekter med tæt relation til aortaklappen (Venturi-effekt).
- subvalvulær pulmonalstenose
- Eisenmenger syndrom ved sen lukning
30.5.4 Atrioventrikulær septumdefekt (AVSD)
Definition og inddeling:
- Komplet AVSD er karakteriseret ved én fælles atrioventrikulær klap med fem flige ('anterior bridging, posterior bridging, right anterior, right posterior, left mural'). Den fælles AV-klap kan indmunde overvejende i den ene ventrikel (ubalanceret AVSD) og dermed gøre den anden ventrikel hypoplastisk (funktionelt univentrikulært hjerte). Chordae kan krydse ventrikelseptum (straddling) og umuliggøre septering (og dermed opnåelse af biventrikulært kredsløb). AV-klappen er som regel insufficient, men sjældent stenotisk.
- Partiel AVSD (= primum ASD): Manglende tilhæftning af atrieseptum til annulus. Der er ledsagende spaltedannelse ('cleft') anteriort i den venstresidige AV-klap. Af og til ses også 'cleft' septalt i den højresidige AV-klap.
- AVSD forekommer hos ca. 35% med Down’s syndrom. Som følge af intensiveret prænatal screening er incidensen af såvel Down’s syndrom som syndromassocieret AVSD faldet. Grundet øget risiko for bevaret høj PVR og dermed manglende symptomer hos børn med Down’s syndrom, foretages der screening neonatalt.
Symptomer:
Komplet AVSD: Børn med store defekter udvikler tidlig hjertesvigt, men kan være asymptomatiske, hvis PVR forbliver høj (risiko for udvikling af Eisenmengers syndrom). Symptombilledet er i øvrigt præget af den underliggende shunt (oftest venstre-højre, men i sjældne tilfælde bidirektionel eller højre-venstre), således funktionsdyspnø, nedsat arbejdskapacitet, cyanose og arytmi.
Partiel AVSD: Ofte ses først symptomer i voksenalderen. Klinik som ved stor sekundum ASD, men afhængig af graden af den venstresidige AV klapinsufficiens (se Afsnit 30.5.1)
Behandling:
Komplet AVSD: Medicinsk hjertesvigtsbehandling samt operation ved tre-måneders alderen med lukning af septumdefekterne og deling af fællesklappen til to AV-klapper. Meget sjældent må pulmonalkredsløbet beskyttes mod et højt flow ved 'banding' af truncus pulmonalis før radikaloperation. For funktionelt univentrikulært hjerte, se Afsnit 30.12.
Partiel AVSD: Operativ lukning og venstresidig AV-klapplastik ved symptomer, hæmodynamisk betydende shunt eller venstresidig AV-klapinsufficiens, typisk i et-årsalderen.
Prognose og opfølgning: Tiårs overlevelse efter operation er over 90%. Der er behov for livslang opfølgning med individualiserede intervaller.
Langtidsfølger:
- AV-klap insufficiens og stenose, specielt venstresidig AV-klap efterlades ofte med nogen insufficiens efter operation
- subvalvulær aortastenose ('goose-neck´ af venstre ventrikels udløbsdel (LVOT))
- progressiv pulmonal hypertension (efter relativ sen AVSD-lukning)
- AV-blok og arytmi
30.5.5 Truncus arteriosus
Definition og inddeling: sjælden tilstand karakteriseret af stor VSD og kun ét fælles arterielt udløb (truncus) fra hjertet, som ovenover truncusklappen deler sig i aorta- og pulmonalarterierne. Der findes flere typer afhængig af pulmonalarteriernes udspring. Truncusklappen er ofte quadricuspid (20%), bicuspid (10%) og/eller dysfungerende (stenotisk eller insufficient), og tilstanden er ofte associeret med abnorm koronaranatomi, bl.a. enkelt koronararterie. Op til 50% har 22q11.2 deletionssyndrom.
Symptomer: Hjertesvigt neonatalt, specielt ved truncusklapinsufficiens. Hvis PVR ikke falder i neonatalperioden, kan børnene forblive asymptomatiske, men i høj risiko for udvikling af Eisenmengers syndrom.
Behandling: Neonatal lukning af VSD og reetablering af forbindelse mellem RV og pulmonalarterierne (indsættelse af homograft eller Contegra-graft).
Prognose og opfølgning: Livslang opfølgning. Næsten alle patienter har behov for reintervention senere i livet, hyppigst pga. stenose og/eller insufficiens af degenereret pulmonal conduit.
Langtidsfølger:
- stenoser sv.t. til proksimale pulmonalgrene
- forkalkning med stenose/insufficiens af homograft mellem RV og pulmonalarterie
- truncusklapinsufficiens (evt. stenose)
- arytmi
- dilatation af aortaroden
- Eisenmengers syndrom
30.6 Venstre ventrikeludløbsdel- og aorta-sygdomme
30.6.1 Valvulær aortastenose
Definition og inddeling: Valvulær aortastenose (se Kapitel 6: Hjerteklapsygdom, Afsnit 6.2) er hyppigste årsag (75%) til udløbsobstruktion, oftest pga. bicuspid aortaklap (0,5-2% af befolkningen, specielt mænd, eventuelt med arvelig NOTCH 1 mutation). Den stenotiske aortaklap kan også være unicuspid, tricuspid, quadricuspid eller dysplastisk. Voksne med bicuspid aortaklap har ofte forstørret annulus, og op til 80% udvikler dilatation af aorta ascendens.
Symptomer: Som ved akvisit valvulær aortastenose. Ved kritisk aortastenose opstår der i neonatalperioden systemisk hypoperfusion pga. ductusafhængig systemcirkulation.
Behandling: Hæmodynamisk betydende stenoser behandles (pallieres) i barnealderen enten med ballondilatation eller kirurgisk valvulotomi. Ved ballondilatation er der risiko for betydende insufficiens og dermed behov for operation i barneårene ofte i form af Ross operation (excision af patientens egen pulmonalklap og indsættelse af autograften i aortaposition samt indsættelse af klapbærende rør (bioprotese) fra RV til pulmonalarterie, typisk homograft eller Contegragraft). Hos udvoksede kan både Ross operation og klassisk klapsubstitution udføres, sidstnævnte oftest med mekanisk klapprotese. Specielt hos yngre kvinder er man tilbageholdende med mekanisk klapsubstitution pga. vanskelighederne med AK-behandling og graviditet. Ikke-opererede voksne patienter behandles som ved akvisit aortastenose (se Kapitel 6: Hjerteklapsygdom, Afsnit 6.2), under hensyntagen til associerede problemer som coarctatio aortae (CoA), aorta ascendens aneurisme (specielt ved bicuspide klapper), eller vedvarende hypoplasi af LVOT.
Prognose og opfølgning: Stort set alle der behandles som børn, får behov for reintervention. Livslang opfølgning for såvel opererede som uopererede børn og voksne. Førstegradsslægtninge (voksne) til patienter med bicuspid aortaklap anbefales screenet med transthorakal ekkokardiografi, hvis to eller flere i familien har kongenit aortaklap sygdom.
Langtidsfølger:
- Ross-opererede udvikler dysfunktion af conduit i pulmonalposition og sjældent insufficiens af autograften i aortaposition (neo-aortainsufficiens).
- Høj frekvens af restenose (15-30%) og tiltagende aortainsufficiens efter ballondilatation. Hvis den primært indsatte klapprotese er lille, kan senere aortaklapsubstitution være nødvendig.
- Risiko for aorta ascendens dilatation/dissektion, specielt ved bicuspid klap og CoA.
30.6.2 Subvalvulær aortastenose
Definition og inddeling: Er sjælden hos nyfødte og betragtes som en progressiv/erhvervet tilstand, som kan skyldes turbulens pga. ændret LVOT-arkitektur. Tilstanden opstår hyppigst pga. en fibrøs ’membran’ eller fibromuskulær ’tunnel’ i LVOT. Den kan også udløses af andre abnormiteter i udløbsdelens strukturer som mitralklappen, chordaes tilhæftning eller ved AVSD (”goose-neck”), men kan også udvikles efter operation for VSD med posterior deviation af udløbsseptum, AVSD, ”double outlet right ventricle” (DORV) og efter switch operation for transposition. Tilstanden er oftest forbundet med progredierende stenose og risiko for udvikling af jet/flow-induceret aortainsufficiens.
Symptomer: Som ved valvulær aortastenose.
Behandling: Symptomatiske patienter opereres. Hos asymptomatiske patienter skal operation overvejes ved middelgradient >40 mmHg og samtidig LV-hypertrofi, svigtende systolisk funktion eller progredierende (selv let) aortainsufficiens. Membranøse obstruktioner reseceres. Ved isoleret tunnelobstruktion udføres resektion af udløbsseptum og evt. modificeret Konno operation med indsættelse af patch i LVOT. Kan kombineres med klapsubstitution eller med Ross operation (Ross-Konno).
Prognose: Lav perioperativ mortalitet. Recidiv hos 20-30% med behov for re-operation hos 15-20%.
Langtidsfølger:
- Risiko for udvikling af aortainsufficiens trods resektion af membran.
- Postoperativt AV-blok.
30.6.3 Supravalvulær aortastenose
Definition og inddeling: Sjælden tilstand med timeglasformet forsnævring sv.t. den sino-tubulære overgang. Ses bl.a. ved Williams syndrom (elastin deletion/mutation i kromosom 7q11.23), som medfører arterielle stenoser med bl.a. udbredt aortahypoplasi, involvering af koronarostier og de store grene fra aorta, såvel som pulmonale grenstenoser.
Symptomer: Som ved valvulær aortastenose. Angina pectoris. Syndromtegn. Risiko/opmærksomhed ved anæstesi pga. mulig uerkendt koronararteriestenose og derfor udtalt følsomhed for tab af systemtryk.
Behandling: Symptomatiske patienter opereres. Asymptomatiske patienter anbefales opereret ved betydende LV-hypertrofi, faldende systolisk funktion af LV, eller hvis koronar- eller aortaklapforhold betinger prognostisk intervention. Hvis aortaklappen kan bevares, udføres udvidelse af de supravalvulære forhold med patch i aorta ascendens eller implantation af rørprotese. Der kan også være behov for intervention på arcus (kirurgi/ballondilatation/stent). Evt. aortarodudskiftning eller Ross type operation.
Prognose og opfølgning: Varierende prognose afhængigt af syndromassociation, grundmorbus og kirurgisk metode. Alle patienter følges livslangt. 20-års overlevelsen efter kirurgi er 80-85%.
Langtidsfølger: Pulmonale grenstenoser regredierer ofte spontant. Øvrige arterielle malformationer kan være progressive. Ca. 25% har vedblivende aortainsufficiens, som dog sjældent progredierer.
30.7 Aortasygdomme
30.7.1 Persisterende ductus arteriosus (PDA)
Definition og inddeling: Persisterende forbindelse mellem aorta descendens og a. pulmonalis. Ofte som led i komplekse hjertesygdomme, hvor kredsløbet kan være 'ductusafhængigt'. Hyppigt forekommende hos præmature.
Symptomer: En stor PDA kan hos børn medføre hjertesvigt, pulmonal hypertension og evt. Eisenmengers syndrom. PDA med lille eller moderat grad af venstre-højre shunt opdages undertiden i voksenlivet som asymtomatisk bifund, årsag til mislyd, venstresidig volumenbelastning, pulmonal hypertension eller lokalisation for IE.
Behandling: Kateterbaseret lukning af PDA anbefales generelt ved hæmodynamisk betydende shunt (Qp/Qs >1,5 eller volumenbelastning af venstre atrium og ventrikel). Lukning foretages ikke som endokarditisprofylakse. Udvikling af Eisenmenger’s syndrom eller desaturation på underekstremiteterne er kontraindicerende for lukning af PDA.
Prognose og opfølgning: God prognose. Ekkokardiografi tre måneder efter lukning og herefter kan patienten afsluttes, såfremt der ikke er restdefekt eller hjertesvigt. Endokarditisprofylakse er indiceret i seks måneder efter ukompliceret lukning
Langtidsfølger: Lille risiko for rekanalisering efter kirurgisk lukning.
30.7.2 Coarctatio aortae (CoA)
Definition og inddeling: Hyppig malformation med stenose på overgangen mellem distale arcus aortae og proksimale aorta descendens, hyppigst distalt for afgangen af a. subclavia sinistra. Kan være kort/lokaliseret eller langstrakt/diffus med betydelig hypoplasi af arcus aortae. Ofte associeret med bicuspid aortaklap (85%), VSD, mitralklapsygdom, cerebrale aneurismer og Turners syndrom.
Symptomer: Svær CoA eller afbrudt aortabue hos nyfødte medfører kredsløbskollaps, når ductus lukker. Mindre defekter giver mislyd og hypertension, men ikke symptomer. Voksne præsenterer sig oftest med hovedpine, svimmelhed, kolde fødder, abdominal angina, claudicatio intermittens og cerebral blødning (cerebrale aneurismer). Klassisk findes blodtryksforskel mellem højre arm og underekstremiteter og svag eller manglende femoralispulse. Diagnosen stilles ved ekkokardiografi og MR/CT.
Behandling: Intervention er indiceret hos voksne med non-invasiv blodtryksgradient >20 mmHg (højre arm til underekstremiteter) med samtidig hypertension (>140/90 mmHg), patologisk blodtrykrespons ved arbejdstest, LV-hypertrofi, betydende aortainsufficiens eller aneurisme af aorta ascendens. Kateterbaseret intervention kan overvejes i fravær af hypertension ved anatomisk svær stenose (50% diameter i forhold til aorta på diafragmaniveau) bedømt ved CT/MR. Interventionsmuligheder er ballondilatation, stentning eller operation (resektion med end-to-end anastomose, indsyning af patch eller rørprotese).
Prognose og opfølgning: 20-års overlevelse over 90%. Livslang regelmæssig opfølgning med billediagnostik og blodtryksmåling i over- og underekstremiteter.
Langtidsfølger:
- arteriel hypertension (hos over 50% trods velbehandlet coarctatio)
- ascendens dilatation/ risiko for dissektions, specielt ved bicuspid aortaklap
- recoarctation, pseudoaneurisme
- aortaaneurisme svarende til end-to-end anastomosestedet eller patchområdet efter aortoplastik
- koronar og cerebral aterosklerose
- endokarditis
- ruptur af cerebralt aneurisme
- hyppigere graviditetskomplikationer (præeklampsi, for tidlig fødsel og maternel hypertension)
For øvrige aortasygdomme henvises til kapitel 11. Aortasygdomme
30.8 Mb. Ebstein og Mb. Ebstein-lignende tricuspidalklapssygdomme
30.8.1 Ebsteins anomali
Definition og inddeling: Apikal displacering af tricuspidalklappen således at den basale del af RV bliver atrialiseret. Septale tricuspidalflig skal være displaceret mindst 8 mm/m² overflade for, at tilstanden betegnes Ebsteins anomali. Oftest optræder samtidig ASD/persisterende foramen ovale (PFO) med mulighed for højre-venstre shunt. Mindre displacering af septale tricuspidalflig forekommer også, hvilket lidt upræcist ofte betegnes som ’mild Ebsteins’ eller ’Ebstein-lignende tricuspidalsklapsanomali’.
Symptomer: Afhænger af displaceringsgraden og tricuspidalklappens funktion (primært insufficiens, men også stenose). Ved svære grader udvikles cyanose i første levedøgn (evt. funktionel pulmonalatresi) pga. højre-venstre shunt gennem ASD/PFO. Svigt af LV (pga. cyanose, dilatation af højre hjertehalvdel, septum abnormiteter, fibrose). Paradoks emboli. Wolff-Parkinson-White (WPW) syndrom er hyppig og ofte med multiple ledningsbaner. Ved milde grader kan der være sparsomme symptomer, også som voksen.
Behandling: Tricuspidalplastik, Cone operation eller klapudskiftning ved insufficient klap. Samtidig lukning af evt. ASD/PFO er indiceret ved NYHA-klasse III-IV, eller ved NYHA-klasse I-II og tiltagende cyanose, højresidigt hjertesvigt eller paradoks emboli. Isoleret ASD-lukning (oftest kateterbaseret) kan overvejes i selekterede tilfælde, men ASD’en er sjældent den væsentligste årsag til patientens symptomer. Ved betydende højre-venstre shunt er der risiko for RV-svigt og nedsat systemisk cardiac output efter ASD-lukning. AK-behandling eller trombocythæmmer overvejes i hvert enkelt tilfælde. Single ventrikel (TCPC) eller 1½-ventrikel strategi (Glenn anastomose) kan vælges ved lille RV.
Arytmi: EKG viser RBBB under normale forhold. Accessoriske ledningsbaner medfører ofte pseudonormalisering snarere end vanligt mønster med delta-tak. Alle typer SVT (fraset atrieflimren) er hyppige, og palpitationer bør føre til henvisning m.h.p. elektrofysiologisk undersøgelse på lav tærskel.
Langtidsfølger:
- hjertesvigt
- arytmier (se Holdningspapir om håndtering af takyarytmi ved kongenit hjertesygdom).
- endokarditis.
30.9 Fallots tetralogi og andre højre ventrikeludløbssygdomme
30.9.1 Fallots tetralogi (ToF) evt. med pulmonalatresi
Definition og inddeling:
- Den hyppigst forekommende form for cyanotisk hjertesygdom (ca. 10% af alle patienter med medfødt hjertesygdom).
- Tetralogien består af en stor perimembranøs VSD, en overridende aorta, pulmonalstenose (infundibulær og evt. valvulær/supravalvulær), og hypertrofisk RV. I ekstreme tilfælde er der ikke forbindelse mellem RV og pulmonalarterierne, hvilket betegnes som pulmonalatresi.
- Bredt spektrum fra milde former med kun beskeden udløbsobstruktion og dermed fravær af højre-venstre shunt og cyanose (pink ToF) til svære grader med lungearterie hypoplasi og pulmonalatresi (ToF med pulmonalatresi eller pulmonalatresi med VSD). I de sværeste tilfælde kan lungeperfusionen være afhængig af ductus arteriosus eller store kollateraler fra aorta direkte til pulmonalarterierne (MAPCA = Major Aorto Pulmonary Collateral Arteries).
- Ca. 15% har mikrodeletion 22q11 (DiGeorge syndrom). Dette er hyppigst forekommende hos patienter med enten højresidig arcus aorta eller pulmonalatresi og MAPCA.
- Associerede læsioner: PFO/ASD, muskulær VSD, AVSD (ofte associeret med Down), højresidig arcus aorta samt koronarkar abnormiteter (hyppigst LAD afgående fra RCA).
Symptomer og objektive fund: Spænder vidt fra ductusafhængigt kredsløb ved pulmonalatresi over varierende grad af cyanose afhængig af sværhedsgraden af pulmonalstenosen til inkompensation ved let pulmonalstenose og dermed udelukkende venstre-højre shunt over VSD’en. Postoperativt oftest EKG med RBBB og evt. bifascikulært blok (15%).
Behandling: Ved svært cyanotiske patienter foretages nogle steder i udlandet tidlig radikaloperation, mens man i Danmark oftest udfører palliation med Blalock-Taussig shunt og senere radikaloperation (staged repair). Hos asymptomatiske foretages i dag typisk radikaloperation omkring seksmåneders alderen, ved shunt-pallierede typisk først i 1-års alderen (alderen ved radikaloperation er faldet løbende gennem de sidste årtier). Operationen omfatter VSD-lukning med en patch og efter behov resektion af infundibulær stenose, valvulotomi, patch til udvidelse af pulmonalannulus (transannulær patch) eller RVOT, samt plastik til udvidelse af stenotiske pulmonalarterier. Ved manglende kontinuitet mellem RV og pulmonalarterierne er der ofte behov for implantation af klapbærende conduit. Reoperation er indiceret ved rest-VSD med betydende shunt (Qp/Qs >1.5), svære stenoser sv.t. pulmonalgrenene, betydende rest-/reobstruktion af RVOT (fx ved RV tryk > 2/3 af systemtrykket), fri pulmonalinsufficiens med tiltagende dilatation (RVESVi) >80mL/m2 og/eller RVEDVi>160mL/m2) eller dysfunktion af RV, nedsat arbejdskapacitet, arytmi og samtidig moderat eller svær tricuspidalinsufficiens. Ved behov for implantation af klapbærende conduit i pulmonalposition benyttes homografter, Contegra-grafter eller biologiske klapproteser. Stentklapper er en mulighed som i tiltagende omfang bruges.
Arytmi: Elektrofysiologisk undersøgelse (EPS) benyttes i stigende omfang til at risikostratificere patienter med risiko for pludselig død (risikofaktorer er LV/RV-dysfunktion, non-sustained symptomatisk ventrikulær takykardi (VT), QRS >180ms, udtalt arvæv på MR). Man bør overveje EPS ved tilstedeværelse af multiple risikofaktorer, ved nærsynkope/synkope eller før kirurgi. Ved inducerbar VT kan ICD i visse tilfælde undlades hos patienter med bevaret LVEF, hvis det relevante VT-kredsløb kan ablateres. I øvrigt tilbydes primær og sekundær profylaktisk ICD på vanlige indikationer.
Langtidsfølger:
- pulmonalinsufficiens (særligt ved transannulær patch men også efter kirurgisk valvulotomi)
- residual eller recidiverende pulmonalstenose (evt. på pulmonalarterieniveau)
- aortainsufficiens evt. med dilatation af aortaroden (15% - trods dette er dissektion sjælden).
- rest-VSD, bilateral ventrikeldysfunktion og endokarditis.
- aneurisme i RVOT.
- arytmier (atrieflagren/flimren, VT).
30.9.2 Pulmonalstenose (intraventrikulære, subvalvulære, valvulære og supravalvulære)
Definition og inddeling: Valvulær stenose skyldes oftest manglende separation af klapfligene, men kan også skyldes dysplastisk fortykkede cuspe (hyppigt ved Noonans syndrom). Muskulær hypertrofi af RVs infundibulum kan medføre dynamisk stenose, nogle gange med meget høje gradienter. Kan også skyldes intraventrikulære muskelbundter. Supravalvulær stenose ses oftest ved Williams syndrom.
Symptomer: Betydende stenoser kan medføre dyspnø, træthed, højresidigt hjertesvigt, arytmi, synkope og brystsmerter. Ved usikkerhed om stenosens betydning for symptomerne, kan det være relevant at foretage ekkokardiografi i forbindelse med arbejdstest, da nogle dynamiske stenoser kan forværres betydeligt ved øget cardiac output.
Behandling: Ved kritisk stenose (ductus-afhængigt kredsløb) neonatalt og sædvanligvis ved peak gradient ved ekkokardiografi >50-60 mmHg (børn og voksne). Næsten alle valvulære stenoser kan ballondilateres (Noonan’s syndrom er den hyppigste undtagelse). Svært dysplastiske klapper, infundibulære og supravalvulære stenoser behandles kirurgisk. Stentklap er en mulighed ved degenereret homograft, Contegra-graft, biologisk protese eller dysfungerende stentklap (valve-in-valve).
Prognose og opfølgning: God prognose. Efter kirurgisk valvulotomi er 50% fri for reintervention efter median på 25 år. Formentlig lavere rate efter ballon-valvulotomi. Ballon-valvulotomi kan gentages ved udvikling af restenose. Livslang opfølgning.
Langtidsfølger:
- restenose
- RV-svigt grundet pulmonalinsufficiens (specielt efter kirurgisk valvulotomi)
- arytmier
30.10 Transposition og double outlet right ventricle
30.10.1 Transposition af de store arterier (d-TGA)
Definition og inddeling: Ved TGA forstås ventrikulo-arterial diskordans dvs. aorta med koronararterier afgår fra RV og truncus pulmonalis fra LV. Det kan være "simpel TGA", eller ved associerede abnormiteter (VSD, CoA, udløbsobstruktion) "kompleks TGA".
Behandling: Ved fødslen er kredsløbet afhængigt af opblanding mellem kredsløbene via ductus arteriosus, en VSD eller mest effektivt gennem en ASD. Nyfødte stabiliseres med prostaglandin for at holde ductus åben, og ved betydende cyanose foretages ballon atrial septostomi (Rashkind) for at øge opblandingen over ASD’en.
Med enkelte undtagelser er voksne med TGA behandlet ved switch operation på atrialt eller arterielt niveau.
Atrial switch operation (Mustard/Senning):
Patienter født før sent i 1980érne er opereret med atrial switch ad modum Mustard eller Senning, hvor blodet redirigeres via intra-atriale tunneller (baffle), hvorved systemveneblodet ledes til LV, hvorfra det løber til pulmonalarterien, og lungeveneblodet ledes til RV, hvorfra det løber til aorta. Langtidskomplikationerne er relateret til RV og AV-klappens tilbøjelighed til med tiden at svigte, så der udvikles hjertesvigt og/eller AV-klapinsufficiens. Der kan opstå problemer med obstruktion eller utæthed i bafflen. Pga. den omfattende kirurgi i atrierne med mange sutur-linjer er der substrat for supraventrikulær arytmi og sinusknudedysfunktion. P-takker er ofte små og svære at få øje på under såvel sinusrytme som arytmi og SVT, specielt atrieflagren, kan vise sig som mindre ændring i PQ-intervallet og hjertefrekvens, men stadig have væsentlig hæmodynamisk betydning.
Arteriel Switch operation:
Verdens første arterielle switch operation blev foretaget i 1975, og behandlingen blev etableret i Danmark i løbet af 1980erne, og anvendes i dag ved de fleste patienter. Ved operationen, som planlægges i første leveuge, deles truncus pulmonalis og aorta ascendens (ovenfor koronararterierne). Koronararterierne eksplanteres med en lille fodplade og reimplanteres i den nye aortarod. Truncus pulmonalis og grene mobiliseres med LeCompte manøver, og de store kar ombyttes og anastomoseres. Prognosen ved arteriel switch er god, da man herved får LV og mitralklap til systemkredsløbet. Langtidsproblemer er sjældne og består i koronarstenose, pulmonalgrensstenose, neo-aortainsufficiens og neo-aortadilatation med klapinsufficiens.
30.10.2 Kongenit korrigeret transposition (ccTGA)
Definition: Sjælden hjertesygdom med (atrio-ventrikulo og ventrikulo-arteriel diskordans) også kaldet ventrikulær inversion. Således modtager morfologiske LV (med mitralklap) blod fra det systemvenøse atrium (morfologiske højre), hvorfra det sendes til a. pulmonalis. Fra det lungevenøse atrium (morfologisk venstre) løber blodet til den morfologiske RV (med tricuspidalklap) som sender blodet til aorta. Herved er kredsløbet serielt forbundet og dermed fysiologisk normalt med fuld iltmætning. Hyppigt ses associerede abnormiteter i form af VSD, pulmonalstenose/-atresi og Ebstein anomali/apikal displacering af tricuspidalklap samt abnormt ledningssystem (AV-blok).
Symptomer og objektive fund: Patienter uden associerede læsioner kan være asymptomatiske til sent i voksenalderen. Patienter født før prænatal hjertescreening blev indført (ca. år 2000), diagnosticeres til tider først som voksne, typisk debuterende med hjertesvigt eller arytmi. Pga. den trabekulerede systemventrikel (morfologiske RV) kan tilstanden forveksles med non-compaction kardiomyopati. Hos voksne er det ikke altid let ekkokardiografisk at definere de fraførende kar, og her er et sikkert tegn at se på AV klapperne. Tricuspidalklappens septale flig hæfter altid nogle mm tættere mod apex end mitralklappen (offset). Samtidig skal der ved en LV kunne erkendes en direkte fibrøs overgang mellem mitralklap og semilunærklap (aorto-mitral curtain). EKG er svært abnormt, da grenbundterne er inverterede sammen med ventriklerne. Herved aktiveres septum fra højre mod venstre, hvorved der ses Q takker i de septale prækordialafledninger. Dette kan mistolkes som tidligere myokardieinfarkt. Præekscitation ses hyppigere, og der er høj risiko for udvikling af forskellige grader af AV-blok.
Behandling: Patienter med associerede defekter vil ofte have undergået intervention i barnealderen (VSD-lukning, ophævelse af pulmonalstenose, system AV-klapprotese). Ved operation ses 50% sandsynlighed for AV-blok, hvorfor mange opererede patienter har pacemaker.
Langtidsfølger:
- højre (system) ventrikel svigt.
- tricuspidalklap (system AV-klap) insufficiens.
- AV-blok (2% /år), enten spontant eller efter hjertekirurgi.
- supra– og ventrikulære takyarytmier (se Holdningspapir om håndtering af takyarytmi ved kongenit hjertesygdom).
30.10.3 Double Outlet Right Ventricle (DORV)
Definition og inddeling: Kompleks sygdom hvor både aorta og a. pulmonalis afgår fra RV. Der er overordnet 4 varianter, som indeles efter VSD’ens lokalisation ift. til de store kar. Subaortal VSD med eller uden pulmonalstenose (Fallot type, VSD-type), subpulmonal VSD (Taussig-Bing anomali) og DORV med fjerntliggende VSD (non-committed type).
Symptomer: Afhængigt af typen og de associerede defekter er symptomerne hos ubehandlede børn cyanose eller VSD-klinik med shunt-betinget hjertesvigt.
Behandling: Kirurgisk korrektion i 1. leveår. VSD-lukning ved DORV med isoleret subaortal VSD. VSD-lukning og samtidig arteriel switch ved isoleret DORV med subpulmonal VSD. I visse tilfælde kan der foretages intrakardiel korrektion uden behov for samtidig switch. Afhængigt af de associerede defekter kan der være behov for Rastelli/Nikaidoh type operation ved DORV med subpulmonal VSD og pulmonalstenose.
Langtidsfølger: Sjældent hos patienter med isoleret subaortal VSD.
- senfølger som hos patienter, som er arteriel switch opereret
- intraventrikulær obstruktion og homograft problemer hos patienter, som er Rastelli opereret
30.11 Tilstande med blivende cyanose i voksenlivet
Definition: Nogle voksne med medfødt hjertesygdom har permanent arteriel desaturation, som forværres ved anstrengelse. Fælles er, at af-iltet veneblod kan løbe direkte til systemkredsløbet eksempelvis via kollateraler til lungevenerne via ASD, eller som hos patienter med Eisenmengers syndrom (se Afsnit 30.11.1) typisk via VSD. Eksempler kan være patienter med Ebsteins anomali, hvor RV-funktionen er for dårlig til, at man kan lukke en ASD. Det kan være patienter med univentrikulære hjerter, hvor man ikke har kunnet lave TCPC-kredsløb, som derfor har balanceret kredsløb (system og lungekredsløb forsynes balanceret fra singleventriklen). Det gælder også for mange TCPC-opererede patienter, at der kan være permanent cyanose pga. tendens til at danne kollateraler fra systemvener til lungevener/venstre atrium.
Permanent cyanose har flere negative konsekvenser, og når man alligevel behandlingsmæssigt vælger at acceptere cyanose, skyldes det typisk en bekymring for om højre hjertehalvdel/ lungekredsløb kan bære et fuldt minutvolumen. Således kan shuntens andel til det systemiske minutvolumen være afgørende for, at cardiac output kan øges. Dette forklarer også, hvorfor anstrengelse forværrer graden af desaturering.
Vedvarende cyanose er en multiorgan sygdom, da cyanose påvirker alle kroppens organer. Der kan ses clubbing af fingre og urglas-formede negle. Der ses hæmatologisk påvirkning i form af sekundær erythrocytose og ofte trombocytopeni, nefropati med mikro/makroskopisk proteinuri og thyroideapåvirkning. Der er en øget forekomst af cerebrale samt pulmonale tromboembolier, cerebrale abcesser, jernmangel og blødning.
Patienter med cyanose kan man møde i alle dele af sundhedsvæsenet. Ved den akutte vurdering hæfter man sig ved, om iltmætningen er som habituelt. En habituelt lav saturation er ikke indikation for iltbehandling, da det sjældent øger saturationen, men primært øger den fysisk opløste O2, og sjældent ændrer på shunt-ratioen. Patientens normale hæmoglobinværdi og hæmatokrit kan meget vel være over laboratoriets normalområde. ”Normale” værdier vil ofte være udtryk for relativ anæmi.
Ved intravenøs behandling af patienter med cyanose skal der bruges luftfilter, da små luftbobler kan give paradoks luftemboli, i værste fald til hjernen. Røntgenundersøgelser med kontrast kan gennemføres uden luftfilter, men med grundig hygiejne for luftbobler.
30.11.1 Eisenmengers syndrom
Definition: Patienter med Eisenmengers syndrom er en særlig undergruppe af patienter med permanent cyanose (kapitel ovenfor). Store, ubehandlede, non-restriktive venstre-højresidig kardielle shunts medfører over få år (typisk 2-3 år) irreversibel skade på lungekredsløbet med fortykkelse af karvæggen og høj pulmonal vaskulær modstand/resistance (PVR) grundet det øget flow igennem pulmonal arterien. Herved bliver det for risikabelt at lukke shuntdefekten. Efterhånden som PVR overstiger den systemiske kredsløbsmodstand opstår højre-venstresidig shuntning og cyanose. Eisenmengers syndrom er en fælles betegnelse for denne fysiologiske kredsløbstilstand, uafhængigt af hvilken underliggende shuntlæsion der er tale om. Incidensen er faldende grundet den forbedrede diagnostik og behandling.
Symptomer og objektive fund: Cyanose, nedsat fysisk funktionsniveau og clubbing af fingre og tæer. Hæmorrhagisk diatese bl.a. med hæmoptyser samt pulmonale og cerebrale tromboembolier. Trods forhøjet hæmatokrit ofte jernmangel og trombocytopeni. Dilatation, aneurismedannelse og dissektion af pulmonalarterierne. Højre- eller evt. biventrikulær hjertesvigt. Supra- og ventrikulære takyarytmier (se Holdningspapir om håndtering af takyarytmi ved kongenit hjertesygdom), endokarditis, cerebral absces, artritis urica, proteinuri og pludselig død.
Behandling:
- Jerntilskud ved jernmangel (transferrin mætning
- Pulmonal vasodilatationsbehandling.
- Antikoagulation kun ved øget risiko for tromboembolisk event såsom atrieflimren/flagren eller lungeemboli.
- Arytmi- og hjertesvigtsbehandling efter vanlige retningslinjer, dog ikke præparater der giver systemiskvasodilatation.
- Venesectio bør generelt ikke foretages, med mindre, det gøres før større kirurgi mhp. at bedre hæmostasen eller ved dekompenseret erythrocytose med moderate/svære hyperviskositetssymptomer (sjældent). Jern mangel/dehydratio skal udelukkes før venesectio, som altid skal foretages i hospitalsregi med tapning af 250-500 ml blod, som successivt erstattes med NaCl.
- Ved indlæggelse anvendes i.v. filtre, for at undgå paradokse luftembolier.
- Ved behov for general anæstesi bør HSE altid konfereres først, da der er risiko for pulmonal hypertensiv krise, hæmodynamisk kollaps og død.
- Transplantation (blok- eller dobbeltlungetransplantation med korrigerende hjertekirurgi).
Langtidsfølger: som ovenfor (se Afsnit 30.11).
30.12 Univentrikulære hjerter – Total Cavo Pulmonary Connection (TCPC)/Fontan-opererede
Definition og inddeling: Fællesbetegnelse for en heterogen gruppe hjertesygdomme, hvor biventrikulært repair ikke er muligt grundet hypoplasi af LV eller RV, eller fordi kredsløbet operationsteknisk ikke kan deles. Eksempler kan være:
- hypoplastisk venstre hjertesyndrom (HLHS)
- double-Inlet left ventrikel (DILV)
- hypoplastisk højre hjertesygdom (HRHS)
- ubalanceret AVSD
Hos mange af disse børn er lungekredsløb eller systemkredsløbet afhængigt af ductus arteriosus (ductus-afhængigt kredsløb), og som nyfødte gives prostacyclin for at holde ductus åben frem til første operation. I modsætning hertil er nyfødte med univentrikulære hjerter med fri blodstrømning gennem pulmonalklap og aortaklap ikke truet ved fødslen. Her udvikles hjertesvigtssymptomer i takt med, at PVR falder og lungerne overperfunderes, og pulmonal banding kan være nødvendigt.
Ved univentrikulært hjerte tilstræber man at opnå TCPC/Fontan Kredsløb. Det foregår ved, at man via en række operationer (2-3) gennem de første 2-3 leveår tilpasser kredsløbet, så ventriklen forsyner systemkredsløbet, mens blodet fra systemvenerne løber passivt til lungerne. Herved opnås serielt forbundet lunge- og systemkredsløb og teoretisk set fuld iltmætning. Lungekredsløbet ved TCPC drives af venetrykket og er afhængigt af lav PVR, og derfor ikke muligt neonatalt. Meget forsimplet er første operation, ud over at være livreddende ved ductus-afhængigt kredsløb, også forberedende til TCPC ved, at man sikrer balance mellem lunge- og systemkredsløb. I 4-6 måneders alderen foretages partiel cavo-pulmonal forbindelse (Glenn operation), hvor overkropsblodet/vena cava superior kobles direkte til pulmonalarterien. TCPC-kompletteringen foretages i 2-3 års-alderen hvor blodet fra underkroppen/vena cava inferior kobles på pulmonalarterien ved hjælp af en gortexconduit.
Hos et fåtal er TCPC ikke muligt pga. øget PVR, og man må i disse tilfælde acceptere, at kredsløbet livslangt er balanceret med cyanose og tidlig udvikling af hjertesvigt.
Mange patienter med TCPC har fin livskvalitet, men langtidsfølgerne er multiple:
- arytmier
- hjertesvigt
- stenoser i Fontan kredsløbet
- valvulopati
- stigende PVR
- kollateraludvikling
- exercise intolerance – manglende evne til at øge minutvolumen målt ved nedsat VO2max
- proteintabende enteropati med ødemer og acites
- venøs/arteriel tromboemboli
- plastisk bronkitis
- levercirrhose
Behandling af langtidsfølgerne omfatter konventionel behandling af arytmi, hjerteklaps operation, kateterbaserede intervention (stenoser i kredsløbet/kollateraldannelse), konventionel behandling af hjertesvigt etc.
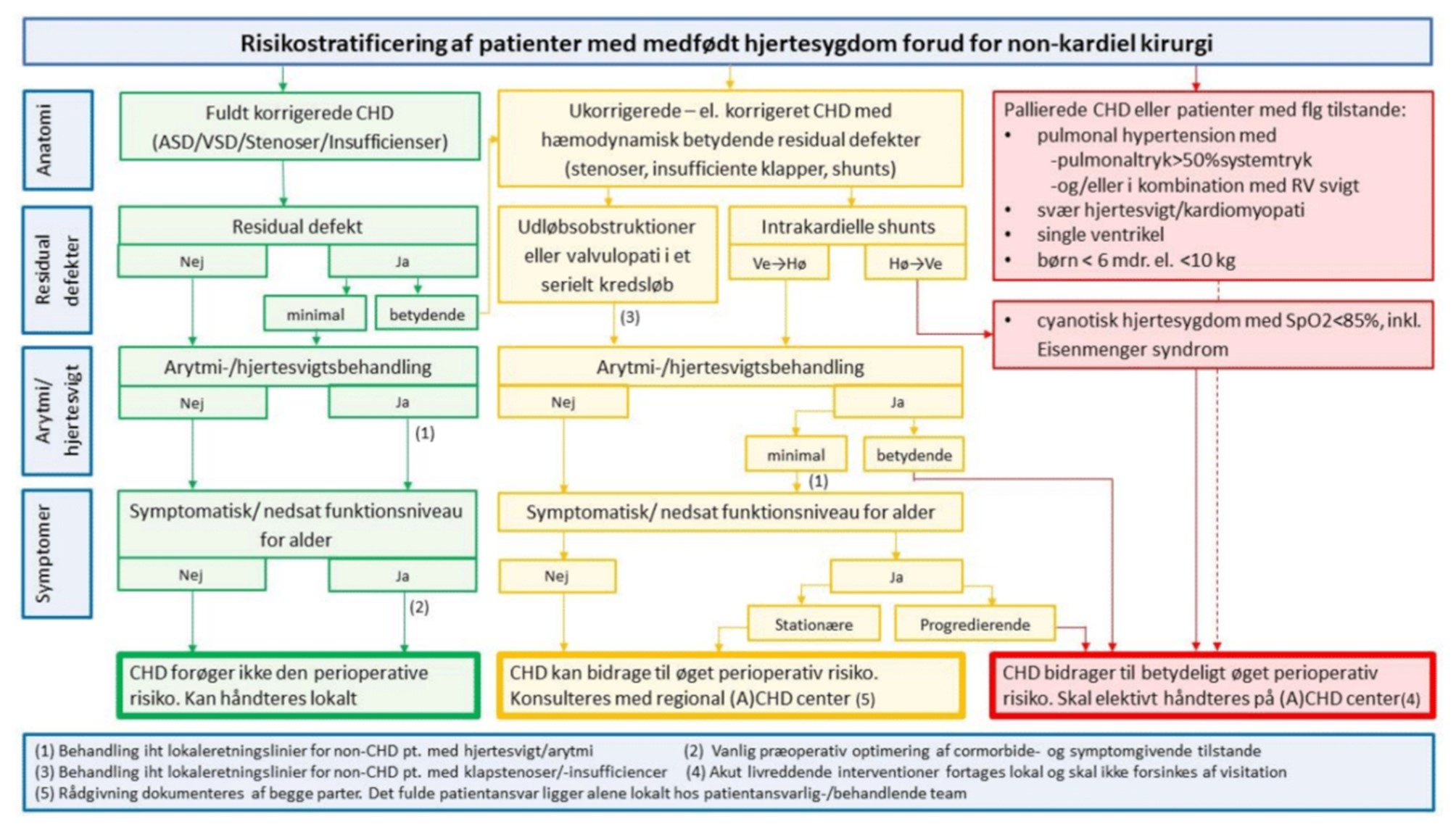
30.13 Flowchart om perioperativ risiko hos patienter med medfødt hjertesygdom
Grundet bedre diagnostik og behandlingsmuligheder bliver populationen med CHD fortsat større og ældre i forhold til tidligere. Grundet dette må fremadrettet forventes et øget behov for kirurgisk/anæstesiologisk håndtering af patienter med CHD.
Organisationsstruktur for behandling af patienter med CHD i DK
Nedenstående flowchart kan anvendes til risikostratificering forud for kirurgiske indgreb. Det må forventes, at ca. 50% af CHD-patienter tilhører ”lav-risiko” gruppen, der kan håndteres med normal anæstesiologisk risiko. De øvrige 50% bør individuelt vurderes i henhold til flowchartet, og rådgivning kan med fordel opsøges på det center, hvor patienter normalt følges.
| Figur 30.1: Flowchart om perioperativ risiko hos patienter med medfødt hjertesygdom |
Bemærk: Ny grafik under udarbejdelse (ingen indholdsmæssige ændringer).
|