32. Arvelige hjertesygdomme


32.1 Generelle forhold
DCS anbefaler systematiseret familieudredning ved en række arvelige eller mistænkt arvelige hjertesygdomme (Tabel 32.1). Nærværende kapitel omtaler kort de enkelte sygdomme samt forhold omkring henvisning, genetisk testning, klinisk opfølgning og juridiske aspekter. For uddybende information henvises til Arvelige Hjertesygdomme, DCS vejledning 2013 nr.1.
Præsymptomatisk udredning af slægtninge motiveres af, at de arvelige hjertesygdomme, f.eks. ionkanal-sygdomme og kardiomyopatier, kan debutere med livstruende arytmi og pludselig død. Andre sygdomme f.eks. familiær hyperkolesterolæmi og dilateret kardiomyopati kan være asymptomatiske, men progressive, og dermed risikeres en irreversibel patologisk udvikling, som potentielt kan forebygges. Ved de fleste arvelige hjertesygdomme foreligger gode behandlingsmuligheder, og tidlig indsats forventes at kunne reducere/forhindre sygdomsudvikling og død iblandt slægtninge.
I klinikkerne for arvelige hjertesygdomme foretages
- undersøgelse og behandling af patienter med arvelige hjertesygdomme
- familieudredning af probander (den først-diagnosticerede i familien) og deres slægtninge
- genetisk undersøgelse af probander og relevante familiemedlemmer
- klinisk opfølgning af slægtninge i risiko for at udvikle sygdom
- risikovurdering af patienter med risiko for ventrikulære arytmier, mhp ICD-implantation
- identifikation af ikke-arvelige differentialdiagnoser
- rådgivning af kollegaer ved klinisk mistanke om arvelige hjertesygdomme hos patienter
32.2 Sygdomme der tilbydes familieudredning for
I Tabel 32.2 ses de diagnostiske kriterier for de hyppigste arvelige sygdomme. For detaljeret information om de enkelte sygdomme, herunder udredning, behandling og kontrolintervaller henvises til:
- Arvelige Hjertesygdomme DCS vejledning 2013 nr. 1
- 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death
- 2023 ESC Guidelines for the management of cardiomyopathies
- NBV Kapitel 8: Myokardiesygdomme
- NBV Kapitel 11: Sygdom i aorta
- NBV Kapitel 18: Ventrikulær arytmi
Tabel 32.2 Diagnostiske kriterier for de hyppigste arvelige hjertesygdomme | ||
Sygdom | Diagnostiske kriterier | Mistænkes ved |
|---|---|---|
Hypertrofisk kardiomyopati | Venstre ventrikel hypertrofi ≥15mm der ikke skyldes hypertension eller klapsygdom (≥13mm i førsteledsslægtninge). | (Lokaliseret) venstre ventrikel hypertrofi uden forklaring. |
Dilateret kardiomyopati | Udelukkelsesdiagnose, som kræver, at der ikke er tale om sekundært nedsat venstre ventrikelfunktion som følge af f.eks. hypertension, klapsygdom, koronarsygdom, længerevarende takykardi m.v. | Uforklaret nedsat systolisk funktion af venstre ventrikel. Arytmiproblematik kan være mere eller mindre fremtrædende. |
Non-dilateret venstre ventrikel kardiomyopati | Uforklaret global/lokal hypokinesi af en ikke-dilateret venstre ventrikel og/eller tilstedeværelse af arvæv eller fedtvæv i venstre ventrikel. | Uforklaret påvirkning af venstre ventrikel uden tilstedeværelsen af dilatation. Arytmiproblematik kan være mere eller mindre fremtrædende. |
Arytmogen højre ventrikel kardiomyopati | Kompleks diagnose, se Arvelige Hjertesygdomme DCS vejledning 2013 nr.1 | Repolaringseringsabnormiteter, dilateret højre ventrikel, VT fra højre ventrikel. |
Langt QT syndrom | QTc≥480 ms eller en sygdomsfremkaldende genetisk variant. Derudover iht Modified Long QT diagnostic score (se ESC 2022 GL). | QTc over 460 ms, og/eller bifasiske T-takker, hos en patient med uforklaret besvimelse, i fravær af reversible årsager eller strukturel sygdom. |
Brugada Syndrom | Spontant type 1 Brugada mønster, set på almindeligt ekg, eller ved høj elektrodeplacering (2. intercostal rum). Type 1 mønster ved medicinsk provokationstest med Ajmaline (tidl. flecainid) kræver yderligere kriterier for at diagnosen stilles, (se ESC 2022 GL). | Ved arytmisymptomer hos patient med strukturelt normalt hjerte, herunder specielt ved polymorf VT. |
Katecholaminerg polymorf ventrikulær takykardi | Bidirektionel VT eller polymorf VT på arbejdstest i fravær af strukturel sygdom. | Besvimelse eller hjertestop i forbindelse med fysisk aktivitet og strukturelt normalt hjerte. |
ST depressions syndrom | Uforklaret ST depression ≥0.1 mV i V3-V6 og/eller I-III. | Uforklarede konstante ST-depressioner, typisk i inferolaterale afledninger. |
Tidlig iskæmisk hjertesygdom | Iskæmisk hjertesygdom med debut | Familiehistorie med iskæmisk hjertesygdom i ung alder (≥2 personer). |
Familiær hyperkolesterolæmi | Se Familiær hyperkolesterolæmi DCS holdningspapir 2019 nr. 2. | Forhøjet LDL kolesterol ≥5mmol/l (≥40 år) eller ≥4mmol/l (18-40 år). |
SCD/aborted SCD | Hjertestop/pludselig død | Hjertestop/pludselig død hvor arvelig hjertesygdom i familien mistænkes. |
32.3 Klinisk udredning lokalt og henvisningsmuligheder
Klinisk udredning lokalt:
Når der diagnosticeres en (formodet) arvelig hjertesygdom, tages der bl.a. ud fra familieanamnesen stilling til, om patienten skal henvises med henblik på familieudredning.
Familieanamnese: Det afklares, om patienten har slægtninge. Herefter vurderes ud fra oplysninger om bl.a. slægtningenes alder, tidlig mulig hjertedød i familien, anden kendt relevant hjertesygdom i familien og komorbiditet, om de kunne have gavn af udredning.
Henvisningsmuligheder:
Der henvises til klinik for arvelige hjertesygdomme i flg. tilfælde:
- alle nydiagnosticerede med en arvelig (eller formodet arvelig) hjertesygdom
- slægtninge til levende eller døde med arvelig eller formodet arvelig hjertesygdom, der skønnes at kunne have gavn af udredning (obs familier med Regionsobduktion varetages af regionsvisitator, se Afsnit 32.8)
- patienter med kendt arvelig hjertesygdom, der overvejer familieforøgelse
Klinisk udredning i klinik for arvelige hjertesygdomme
I klinikken for arvelige hjertesygdomme vurderes den sandsynlige arvegang, penetrans og ekspressivitet ud fra patientens fænotype og familieanamnese (baseret på stamtræs-optegnelse). Det fastlægges dermed hvilke familiemedlemmer, der er i risiko for sygdomsudvikling, og om der er grund til at tilbyde udredning af andre end 1. leds-slægtningene. Herefter kontaktes slægtningene. Den indledende kontakt til slægtningene foregår som hovedregel gennem probanden vha. et familieskema. Efter modtagelse af tilsagn fra den enkelte slægtning, tilbydes personen samtale om udredningens formål, indhold, mulige betydning mv. Der tilbydes herefter et sygdomsspecifikt klinisk udredningsprogram (Tabel 32.3).
Klinisk udredning af probanden forudgås som hovedregel af genetisk undersøgelse. Hos slægtninge foregår den kliniske og genetiske udredning ofte parallelt. Dette motiveres bl.a. af, at behandling primært baseres på symptomer og kliniske fund, samt at den kliniske betydning af genetiske fund ofte skal fortolkes i forhold til kliniske fund i familien. Desuden behandles og følges de familier, hvori der ikke påvises en genetisk variant i det anvendte genpanel, ud fra kliniske fund alene.
Tabel 32.3: Initielt udredningsprogram af slægtninge ved mistanke om arvelig hjertesygdom | ||||||||
| Anamnese, Klinisk Us | EKG BP | EKKO | Holter | Sene pot. | Arbejdstest | Risiko score | MR |
|---|---|---|---|---|---|---|---|---|
Hypertrofisk kardiomyopati | X | X | X | (X) | (X) | (X) | ||
Dilateret kardiomyopati | X | X | X | (X) | (X) | |||
Non-dilateret venstre ventrikel kardiomyopati | X | X | X | (X) | X | |||
Restriktiv kardiomyopati | X | X | X | (X) | ||||
Arytmogen højre ventrikel kardiomyopati | X | X | X | X | (X) | (X) | (X) | |
Muskeldystrofi | X | X | X | X | ||||
Langt QT-syndrom | X | X | Xb | (X) | ||||
Kort QT syndrom | X | X | Xb | (X) | ||||
Brugada syndrom* | X | Xa | Xb | (X) | ||||
Katecholaminerg polymorf VT | X | X | Xb | X | X | |||
Familiær ST-depression syndrom | X | X | X | X | (X) | (X) | ||
Tidlig repolarisations-syndrom | X | X | Xb | X | ||||
Kronisk progressiv overledningsforstyrrelse | X | X | X | X | ||||
Tidlig iskæmisk hjertesygdom | X | X | X | |||||
Familiær hyperkolesterolæmi | X | X |
| |||||
Transtyretin amyloidose** | X | X | X | (X) | ||||
Fabry sygdom | X | X | X | (X) | ||||
Pulmonal arteriel hypertension | X | X | X | |||||
Arvelige thorakale aorta-sygdomme*** | X | X | X | X | ||||
Pludselig uventet hjertedød | X | Xa | X | X | (X) | |||
Idiopatisk VF | X | Xa | X | X | (X) | |||
X=undersøges hos alle; (X)=evt. suppl. undersøgelse afhængig af resultatet af initiale undersøgelse. Xa inkl. løftet elektrode placering. Xb udføres initielt differentieldiagnostisk eller hvis det hos patienten skønnes relevant inden start af betablokade. *Ved mistanke om Brugada syndrom anvendes ajmaline-test (tidligere flecainid) med løftet elektrode placering som led i udredningen. **Ved mistanke om transtyretin amyloidose foretages DPD skintigrafi. ***De syndrom-associerede aortasygdomme udredes iht. et særligt cirkulære i Center for Sjældne Sygdomme på Aarhus Universitetshospital og Rigshospitalet – de øvrige i kardiologisk regi. Ved non-syndrom associeret arvelig øget risiko for thorakal aortasygdom tilbydes henvisning til familieudredning i det kardiologiske center. ****Ved pludselig død (Pludselige uventede dødsfald under 50 år DCS vejledning 2013 nr. 2) samt Afsnit 32.8 Regionsobduktion. En udredning af slægtningene kan være sygdomsspecifik eller mere omfattende, i fald obduktion er blank. | ||||||||
32.4 Genetisk testning og rådgivning
Den genetiske udredning foretages primært af kardiologer i klinikker for arvelige hjertesygdomme og danner, afhængig af sygdomsenhed, i forskellig grad basis for vurdering af prognose, behandlingsmuligheder og behov for profylaktiske tiltag samt prænatal diagnostik. Sidstnævnte varetages oftest af klinisk genetisk afdeling. Den genetiske udredning foregår i samarbejde med kliniske genetikere, patologer, retsmedicinere, pædiatere, molekylærbiologer samt biokemikere. Af Tabel 32.1 fremgår, hvilke sygdomme, der aktuelt kan tilbydes genetisk testning for.
Formålet med genetisk testning er at be- eller afkræfte bærerstatus for en sygdomsfremkaldende variant med henblik på at kvalificere behov for opfølgning, profylaktiske tiltag eller prænatal diagnostik.
Som udgangspunkt undersøges probanden bredt for varianter i gener, der er kendt at kunne give ophav/disponere til den pågældende sygdom. Ved den bioinformatiske analyse graderes de fundne varianterne fra 1 til 5, hvor klasse 4 (likely pathogenic) og klasse 5 (pathogenic) varianter som udgangspunkt betragtes som værende sygdomsfremkaldende. Blandt slægtningene undersøges specifikt kun for den/de sygdomsfremkaldende varianter (klasse 4 og 5), der er fundet hos probanden. Ved fund af klasse 3 varianter (variant of unknown significance [VUS]), kan slægtningene undersøges for at vurdere kosegretion i familien (dvs. de individer, der bærer varianten har også fænotypisk sygdom hvorimod de individer der ikke bærer varianten er raske). Derved kan man komme nærmere hvorvidt varianten reelt er sygdomsfremkaldende, men den kan ikke bruges til at udelukke risiko for at udvikle sygdommen. I alle tilfælde vil tilstedeværelsen af en sygdomsfremkaldende variant hos probanden have betydning for, hvordan slægtningene udredes.
Kardiologen i klinik for arvelige hjertesygdomme varetager hovedparten af den genetiske rådgivning, men i en række tilfælde kan der være behov for rådgivning ved klinisk genetiker for eksempel ved mistanke om syndrom, prænatal diagnostik, præimplantations-genetisk diagnostik (= ægsortering), diagnostik af ikke myndige eller efter patientønske.
De mulige udfald af gentestning og anvendelsen af svarene er illustreret i Figur 32.1.
Figur 32.1: Genetisk testning og anvendelse af svaret til udredning af slægtninge |
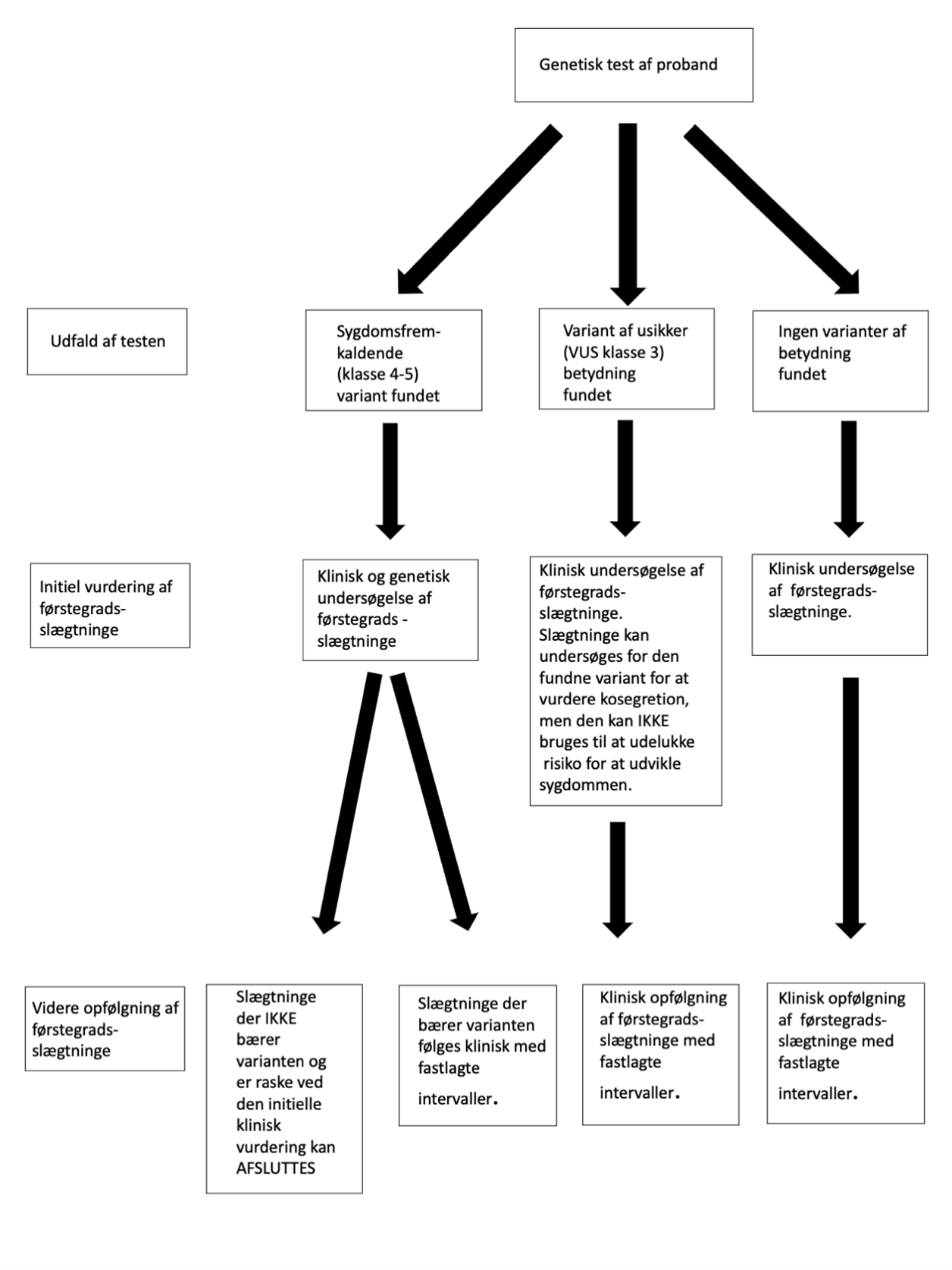 |
VUS = Variant of unknown significance |
Udredning af børn
Det anbefales almindeligvis ikke, at børn undersøges for sygdomme, der helt overvejende debuterer efter den alder, hvor barnet selv kan tage stilling (15-18 år). Ses den pågældende sygdom imidlertid i barnealderen (som for eksempel ion-kanalsygdomme, familiær hyperkolesterolæmi og hypertrofisk/dilateret kardiomyopati) anbefales klinisk undersøgelse, i samarbejde med pædiaterne. Genetisk undersøgelse af børn anbefales efter individuelt skøn, men som hovedregel kun såfremt et positivt (eller negativt) svar a priori skønnes at munde ud i aktiv behandling (eller ophør heraf) i barnealderen (f.eks. ved langt QT-syndrom eller katecholaminerg polymorf ventrikulær takykardi). For uddybende information henvises til Arvelige hjertesygdomme hos børn DCS vejledning.
32.5 Klinisk opfølgning af slægtningene efter udredningen
Proband har sikkert sygdomsfremkaldende variant:
Syge slægtninge, der får påvist sygdom, følges og risikovurderes som probanden.
Slægtninge, der ikke har familiens sikkert sygdomsfremkaldende genetiske variant, afsluttes.
Slægtninge der er raske bærere af en sikkert sygdomsfremkaldende genetisk variant følges i et sygdomsspecifikt kontrolprogram.
Proband har ingen sikker sygdomsfremkaldende variant:
Syge slægtninge, der får påvist sygdom, følges og risikovurderes som probanden.
Raske slægtninge uden eller med kun få symptomer og uden eller med borderline objektive fund, følges med individuelt tilrettelagte intervaller, idet de er i risiko for at udvikle manifest sygdom senere i forløbet.
32.6 Juridiske aspekter
Slægtninge, der tilbydes udredning, skal forud for klinisk konsultation grundigt informeres om, at hvis der findes tegn på sygdom, kan det få betydning for fremtidig tegning af pensioner og forsikringer, erhvervsvalg (herunder sport) og ved vurdering af egnethed til adoption. Genetiske fund og normale kliniske undersøgelsesresultater opfattes at være omfattet af tavshedspligten og utilgængelige for tredje part, herunder forsikringsselskaber. Forsikringsselskabet må ikke spørge, om der er sygdom i familien. À priori raske slægtninge over 18 år, kan før de indkaldes til undersøgelse i hjerteambulatoriet, tegne en forsikring eller pensionsordning på helt samme vilkår og betingelse som enhver anden hjerte-rask person. Det samme gælder raske slægtninge, der er i kontrolforløb.
32.7 Nationalt Genom Center
Som led i Regeringens og Danske Regioners målsætning om at styrke personlig medicin i Danmark, herunder brug af genetiske data i diagnostik, behandling og forskning, blev Nationalt Genom Center (NGC) etableret i 2019 som en selvstændig styrelse under Sundhedsministeriet. NGC’s infrastruktur skal sikre patienter, læger og forskere lige adgang til hel-genom sekventering (WGS) og opbevaring af oplysningerne i en National Genom database på tværs af landet. Nogle af de arvelige hjertesygdomme er udvalgt som et af de første sygdomsområder, der tilbydes WGS i NGC regi.
Siden 1. juli 2019 har det været et lovkrav, at patienterne skal afgive skriftligt samtykke ved omfattende genetisk analyse og tage stilling til i hvilken udstrækning, de vil informeres om sekundære fund (se Nationalt Genom Center: samtykkeblanket til patienter). De har også ret til at bestemme, at deres genetiske data, der opbevares i Nationalt Genom Center, ikke må anvendes til sundhedsforskning. Ovenstående betyder også, at det for nuværende ikke er muligt at foretage omfattende genetiske analyser på afdøde i NGC regi.
Information og genetisk rådgivning af patienten, blodprøvetagning samt indhentelse af samtykke foregår i de enheder, der varetager udredning og behandling af arvelige hjertesygdomme. DNA-sekventering og analyse for mindre og større strukturelle varianter foretages i et af de to nationale sekventeringscentre. Variantfortolkning foretages af de henvisende centre på NGC’s computer med adgang til fortolkningsværktøj inde på NGC’s computer samt til kliniske data fra de henvisende afdelinger, hvorefter der udarbejdes en rapport til den rekvirerende kliniske afdeling.
32.8 Regionsobduktion
Siden 2021 er der skabt mulighed for at rekvirere obduktion af uventet og pludseligt døde ved de retsmedicinske institutter i de tilfælde, hvor politiet ikke ønsker obduktion. Denne nye ordning benævnes regionsobduktioner. Det overordnede formål med regionsobduktioner er at foretage obduktion af en person, som pludseligt og uventet er afgået ved døden med henblik på at afdække – eller udelukke - eventuel arvelig sygdom som dødsårsag, og dermed få mulighed for, at der på bedst muligt grundlag evt. kan tilbydes forebyggende kontrol og behandling af de pårørende. Hvis politiet ikke ønsker en retslægelig obduktion, skal behandlende læge henvise til regionsobduktion/konferere med regionsvisitator, hvis der er mistanke om arvelig sygdom.
Arbejdsgangen i forbindelse med udarbejdelse af dødsattest i alle tilfælde af pludselig og lægefaglig uventet død er nu således, at behandlende læge skal:
- kontakte politiet mhp indikation for retslægeligt ligsyn og obduktion
- udfylde dødsattesten (side 1)
Hvis politiet ikke finder indikation for retslægelig obduktion (evt. efter retslægeligt ligsyn som ikke har medført obduktion), da informere pårørende om muligheden for regionsobduktion, og at indikation for obduktion vil blive drøftet med regionsvisitator i tvivlstilfælde, før der vil blive taget endelig stilling til, om det er relevant at foretage obduktion. Regionsvisitator kontaktes i henhold til gældende instrukser for den enkelte region.
- indhente samtykke fra pårørende til mulig regionsobduktion
- henvise afdøde (hvor der foreligger samtykke) under 50 år direkte til regionsobduktion til retsmedicinsk institut og for afdøde over 50 år eller i tvivlstilfælde til visitator for regionsobduktioner på sikker-mail eller ved telefonisk kontakt.
Henvisningen skal indeholde udfyldt skema (link eller direkte i elektronisk patientjournal afhængig af region) med information om dødsfald (evt. udskrift fra ambulancejournal eller AED), medicin, komorbiditet og familiære disposition til pludselig død. Side 1 fra dødsattesten sendes med.
Svar på obduktionen vil kunne ses i journal og på sigt i Patobank under afdødes CPR nummer.
Hovedansvarlig regionsvisitators mailadresse (ikke til akut visitation)
Region Nordjylland: regionsobduktion@rn.dk
Region Midtjylland: auh.hjertesygdomme.regionsobduktion@rm.dk
Region Syddanmark: ouh.ode.b.arvamb@rsyd.dk
Region Sjælland: regionsobduktion@regionsjaelland.dk
Region Hovedstaden: regionsobduktion.rigshospitalet@regionh.dk
Mail til retsmedicinske institutter
København: rpa@sikkermail.forensic.ku.dk
Odense: ri@sdu.dk
Aarhus: sikker@forens.au.dk